
Összefoglaló közlemények / Reviews
Microbiome Testing Methods and Challenges of Clinical Interpretation
Summary
It is a great challenge for practitioners not having internationally uniform guidelines available for the methodologies of microbiome testing and evaluating results. While on the internet there is a rapidly growing amount of preclinical and clinical data describing clear disease associations in randomised controlled trials readily accessible to patients, guidelines for clinical decision-making algorithms at the bedside are currently lacking. Our goal is to compare different microbiome testing methods and to shed light on the biological significance of the most frequently appearing parameters in the findings: diversity, phylogenetic and functional taxonomic groupings (e.g. the F/B ratio) and its clinical relevance. The clinical application must integrate the diagnostic and therapeutic approaches of several fields of expertise with completely new and dynamically developing molecular diagnostic and nutritional therapeutic guidelines. It is imperative for the specialised physician managing the patient's treatment to receive professional help from molecular biologists, microbiologists, and related professions when evaluating the findings and making a therapeutic decision. The microbiome is typically associated with the human body on the barrier surfaces of the alimentary canal, such as the mouth, gastrointestinal tract and the skin, etc., so it is essential to involve specialists and imaging disciplines as early as biopsy sampling and/or treatment. According to our recommendations, interdisciplinary teams similar to tumour boards are needed for the successful clinical application of the results.
Összefoglalás
A gyakorló klinikus számára nagy kihívást jelent, hogy a mikrobiom vizsgálatának és értékelésének módszertanára nincs nemzetközi szinten egységes irányelv. Miközben az interneten a betegek számára is elérhető, robbanásszerűen növekvő számú preklinikai és klinikai adat egyértelmű betegségasszociációkat ír le randomizált kontrollált vizsgálatokban, a betegágy melletti döntési algoritmusok irányelvei egyelőre hiányoznak.
Célunk a mikrobiom vizsgálati módszereinek összehasonlítása, illetve a leletekben leggyakrabban megjelenő paraméterek: a diverzitás, a filogenetikai, illetve funkcionális taxonómiai csoportosítások (pl. az F/B arány) biológiai jelentőségének és klinikai relevanciájának megvilágítása.
A klinikai alkalmazásnak több szakterület diagnosztikus és terápiás szemléletét kell integrálni teljesen új és dinamikusan fejlődő molekuláris diagnosztikai, illetve táplálásterápiás irányelvekkel. Fontos, hogy a beteg kezelését irányító szakorvos a leletek értékelésekor és a terápiás döntés során szakmai segítséget kapjon a molekuláris biológusoktól, mikrobiológusoktól és a társszakmáktól. A mikrobiom jellemzően a tápcsatorna, a száj és a bőr stb. barrierfelületein van az emberi szervezettel kapcsolatban, ezért már a mintavétel és/vagy a kezelés során ebben jártas szakorvosok, képalkotó diszciplínák bevonása elengedhetetlen.
Javaslatunk szerint ezért az onkoteamekhez hasonló, összeszokott interdiszciplináris teamekre van szükség az eredmények sikeres klinikai alkalmazásához.
Bevezetés
A humán mikrobiom az emberi szervezettel együtt élő baktériumok, gombák, archeák és vírusok összessége.
Az emberi mikrobiommal kapcsolatos, egyre bővülő ismeretek minden bizonnyal a XXI. század meghatározó orvostörténeti eseményei között lesznek majd. Abban mindenki egyetért, hogy a jövőt a mikrobiomasszociált betegségek táguló értelmezési szempontjai meg fogják határozni. Az utolsó 10 év robbanásszerű bővülést hozott a rendelkezésre álló ismeretanyagban, ugyanakkor a vizsgáló módszerek párhuzamos fejlődése beláthatatlan távlatokat nyitott az eredmények értelmezésében. A jövőkutatók perspektívájából ez biztató, de az internet korában, gyakorló orvosként azzal szembesülünk, hogy a tudományos közlemények áradatában még egységes nómenklatúra sincs. Hol húzzuk meg a baktériumfajok határait? Mit tekintünk azonosított fajnak? Mi alapján számoljuk a fajgazdagságot? Egyelőre úgy tűnik, hogy ezek a nevezéktani disszonanciák és a „nagyobb felbontású”, részletgazdagabb vizsgálati módszerek nem segítenek csökkenteni, hanem inkább növelik a természettudományokban máshol megszokott bizonytalanságot (pl. Heisenberg-féle határozatlanság). Ezt elméleti síkon könnyebb kezelni, mint gyakorló klinikusként. Tapasztalt klinikusi pragmatizmusra van tehát szükség, ahol a releváns eltérések asszociációját vizsgáljuk valós klinikai problémákkal, és ezeket elkülönítjük a kockázatot nem jelentő eltérésektől. A napi gyakorlatból vett példával élve, ha egy páciens megsemmisítő mellkasi fájdalommal érkezik, és ennek hátterében akut koronáriabetegség határozottan kizárható, akkor a magas koleszterinszintje még akkor sem bír jelentőséggel a panaszai szempontjából, ha hosszú távon fontos kóroki tényező és kockázat, amit kezelni és kontrollálni kell. Ugyanakkor a beteg akut panaszait refluxbetegség vagy pszichoszomatikus tünetképzés vezérli, és ilyen irányú célzott kezelésre szorul.
A közlemény célja a gyakorló klinikus számára értelmezhetővé tenni a mikrobiomdiagnosztika alapjait, annak metodikai és interpretációs aspektusával és bizonytalanságaival. A klinikai döntés szempontjából ugyanis talán nagyobb jelentősége van annak, hogy pontosan értsük azt, amit nem tudhatunk meg egy vizsgálatból, mint azt, amit esetleg igen. Egy újabb gyakorlati példával élve és a fenti esetnél maradva egy nyugalmi EKG nem zár ki fokozott infarktuskockázatot. Ehhez legalább kísérő laborok, terheléses EKG, echokardiográfia kell, szükség esetén kardio-CT vagy koronarográfia. Ettől még a nyugalmi EKG egy fontos vizsgálati eszköz, azonban minden klinikai diagnosztikai eljárás értékét, hasznosságát, „használhatóságát” a klinikum dönti el. Mi a kérdésfeltevés? Ehhez lesz illeszthető egy adott vizsgálóeljárás érzékenysége, specificitása és költsége. Az előző példánál maradva egy 1 hetes Holter-monitor az EKG-mérés rendkívül korszerű formája, de hasznossága erősen kérdéses, ha akut mellkasi fájdalomról van szó, nem beszélve arról, ha az értelmezéshez használt szoftver bizonytalansága olyan mértékű, hogy a 168 óra adat automatikus értékelése, mondjuk, 50 óra humán elemzést feltételez. Tehát egyrészt az adatgyűjtés mélysége kérdéses, hogy releváns-e a klinikai kérdés szempontjából, másrészt az, hogy a kiértékelésre használt szoftver gyorsasága, elemzőkészsége, megbízhatósága, pontossága megfelelő-e a válaszadáshoz.
A mikrobiomdiagnosztika
A mikrobiomasszociált (nem fertőző) betegségek „prototípusa” a reumás láz. Klinikai tünettanát Sydenham a 17. században már jóval a bakteriológia hőskora előtt leírta, a Streptococcusok kóroki szerepének azonosítása (és elnevezésük is) ugyanakkor az osztrák sebészóriás, Theodore Billroth nevéhez fűződik (1). A következő kb. 100 évet meghatározta a mikroszkópos morfológiai diagnosztika és a tenyésztéses technológiák fejlődése. Újabb forradalmi lépést a kóroki tényezők célzott eliminációjának lehetősége hozott az antibiotikumok megjelenésével. Ez alapjaiban változtatta meg a népegészségügy prioritásait: a szifilisz, a tbc, az endocarditis mellett a reumás láz okozta morbiditás és mortalitás is meredeken zuhanni kezdett – az 1920-as években az 5-20 év közötti korcsoportban a vezető halálok volt a fejlett világban, és heteket, hónapokat töltöttek az érintett páciensek rehabilitációs intézetekben (2).
A mikrobiomkutatás Nobel-díjat érő forradalmi mérföldköve, a Helicobacter pylori felfedezése is egy új diagnosztikus megközelítés eredménye volt. Barry Marshall és Robin Warren korszakalkotó megfigyelése a patológiai rutindiagnosztika eszköztárával tette láthatóvá a Gram-negatív, spirális pálca alakú baktériumokat, ami forradalmi változást hozott az atrófiás gastritis, a fekélybetegség, a gyomorrák és a MALT lymphoma megelőzésében és kezelésében (3). Ez felhívta a figyelmet arra, hogy a gasztrointesztinális traktusban élő, rutinszerű tenyésztéssel ki nem mutatható baktériumok jelentős kóroki szerepet játszhatnak addig ismeretlen eredetű gyulladásos, degeneratív és daganatos megbetegedésekben egyaránt. Ez különösen annak fényében fontos, hogy az emberiség kb. 40%-a Helicobacter-kontaminált, (4) és a betegségasszociáció ennek fényében multifaktoriális kell hogy legyen. Másrészt felhívta arra is a figyelmet, hogy a Helicobacter-negatív esetekben is gondolnunk kell baktériumok kóroki szerepére, hiszen magának a Helicobacternek a kimutathatósága a kor diagnosztikus lehetőségeivel esetleges (és véletlen) volt.
A genomika térhódítása, illetve a humángenom-projekt által generált technológiai robbanás – az ún. új generációs szekvenálási eljárások megjelenése – hozta el a technológiai áttörést, ami megfelelő vizsgálómódszernek bizonyult az emésztőtraktus baktérium- (illetve gomba-, archea- és vírus-) populációjának vizsgálatához (5). A genetikai taxonómia úttörői, Carl Woese és munkatársai az 1980-as években azonosítottak a baktériumgenomban olyan szakaszokat, amelyek alapján filogenetikai kapcsolat volt megállapítható az egyes fajok, nemzetségek között (6). Ez a baktériumriboszómát kódoló DNS-szakasz kellően konzervált volt az egyébként meglehetősen dinamikus evolúciót mutató bakteriális genomon belül. Szemben a bakteriális enzimeket kódoló génekkel, amelyek evolúciós alkalmazkodást nemzedékek között is gyorsan lehetővé tesznek (pl. a laktáz enzim mutációja nyomán alternatív anyagcserére is átállhat a baktérium), a riboszóma-RNS annyira alapvető funkciót kódol, hogy genetikai állománya rendkívül lassú evolúcióra képes csak, így egyfajta kronométerként a törzsfejlődési kapcsolat azonosítását lehetővé teszi. Az egyedek közötti minél nagyobb különbség filogenetikai „távolságot” jelöl. Woese és munkatársai ezzel a szemlélettel azonosították elsőként az archeákat különálló doménként a prokarióták és eukarióták mellett.
A prokariótákban az S16 szakasz 1500 bázispár (bp) hosszúságú, de technikai okok miatt sokáig az első 500 bp hosszúságú szakasz alapján történt a fajta és diverzitás meghatározása. Az első ilyen megközelítések a szekvenciaadatokat referencia-adatbázisokkal összevetve határozták meg az egyedi taxonómiai egységeket, amelyek genetikai homogenitását konszenzusos alapon 97%-os azonosságnál húzták meg. Ez alapján lehet közelíteni a faj- és nemzetségmeghatározásokhoz is, amelyet azonban mind a módszertan (a 16S gén 1500 bp hosszúságához képest mért tényleges fragmenshossz), mind az azonosításhoz alkalmazott szoftver, illetve maga a referencia-adatbázis kritikusan befolyásol (7).
Mint minden teszt esetében, itt is igaz az, hogy minél hosszabb szakaszt akarunk mérni, annál pontosabb a meghatározás specificitása, de biológiai mintáról lévén szó (jellemzően széklet, esetleg fixált szövet) nagy a DNS/minta fragmentáltság mértéke, ami csökkenti az analitikai szenzitivitást. Ugyanakkor minél rövidebb a vizsgált génszakasz, annál könnyebb a tesztet reprodukálhatóan elvégezni, viszont annál kisebb az eredmény specificitása. A technológia fejlődésével egyre inkább egyeduralkodóvá válik az S16 szakasz teljes hosszának vizsgálata, ami a mai tudásunk szerint megfelelő pontosságú faj- és nemzetségmeghatározást tesz lehetővé (8).
Látni kell ugyanakkor, hogy a filogenetikai klasszifikáció előnye egyben a hátránya is, nevezetesen az, hogy a faj azonosítása mellett az adott baktériumok tényleges enzimkészletéről, toxint termelő képességéről, rezisztenciaviszonyairól nem ad információt. A rákgenom és a molekulárisan célzott gyógyszerek kapcsolata, és ennek prediktív diagnosztikai alkalmazása révén tudjuk ugyanakkor, hogy még agy adott (igazoltan) kóroki szerepet játszó gén jelenléte sem jelent azonos klinikai fenotípust. Egészen pontosan ismernünk kell egy molekuláris célpontot definiáló gén szekvenciájának és a kódolt fehérje térszerkezetének összefüggéseit ahhoz, hogy a funkcióra következtetni lehessen. Vannak olyan mutációk ugyanis, amelyek aminosavcsere vagy deléció/inszerció révén funkcionális konzekvenciát jelentenek, míg mások nem. A gyógyszerkötőhelyek konformációváltozásai kapcsán ez az affinitást és ezen keresztül a hatékonyságot is befolyásolja (9).
Nem ismeretlen ez a dilemma a konvencionális mikrobiológiai diagnosztikában sem. Általános követelmény egy diagnosztikai eljárással szemben, hogy a klinikailag releváns kérdés tekintetében kellően érzékeny, specifikus és költséghatékony legyen. Épp ezért a klinikus feladata, hogy olyan diagnosztikai módszert válasszon, amelynek terápiás konzekvenciája van, és költséghatékony.
A Helicobacter-diagnosztika esetében a standard hematoxilin- és eozinfestés mellett érzékenyebb és specifikusabb immunhisztokémiai technikák is elérhetővé váltak (10), illetve megjelentek a prediktív molekuláris genetikai tesztek a patogenitásfaktorok, illetve gyógyszer-rezisztenciáért felelős gének tekintetében (11). De mikor melyiket alkalmazzuk? Hogy befolyásolta ez a diagnosztikai algoritmusokat? Egy szövődménymentes gastritis esetében elég lehet szövettan vagy kilégzési teszt, hiszen elég kimutatnunk a baktérium jelenlétét, az első vonalas gyógyszerválasztáshoz szakmai protokollok vannak, nem szükséges további molekuláris karakterizálás. A MALT lymphoma kezelése előtt ugyanakkor az esetleges kombinált kezelés, illetve már az első vonalas gyógyszerválasztás miatt fontos ezt kiegészíteni további molekuláris markerekkel, ami a patogenitás és a gyógyszerérzékenység, antibiotikumrezisztencia szempontjából a malignus betegség miatt célzottabb megközelítést igényel.
Fontos szempont ezek mellett a terápiás beavatkozás kockázata. Ez a mikrobiom tekintetében a legtöbb esetben nem maga az intervenció, hanem a helytelen differenciáldiagnózis miatti helytelen terápia és az időveszteség, amely a betegség progresszióját lehetővé teszi. Fel nem ismert Klacid-rezisztencia esetén az előbbi példánál maradva még egy gastritis esetén is jelenthet hiábavaló antibiotikus kezeléseket vagy hosszú távú protonpumpagátló-kezelést. Ezért ha első vonalban nem volt sikeres az eradikáció, a második vonal terápiaválasztása előtt mindenképp felmerül a baktérium pontosabb molekuláris karakterizálása.
Egy másik példán érzékeltetve a módszertani dilemmát: a Clostridium (új filogenetikai klasszifikáció szerint Clostridiodes) difficile fajok patogenitása a toxintermelő képesség függvénye. A konvencionális tenyésztés mellett ezért egyre inkább teret nyer a patogenitással és virulenciával szoros(abb) összefüggésben lévő toxinok, illetve az ezeket kódoló gének kimutatása különböző technológiákkal. Többféle toxint kódoló gén (tcdA+, tcdB+, cdtA+, cdtB+) és ezek kombinációi, illetve az antibiotikumrezisztenciát kódoló gének is kimutathatók, ami egészen precíz és személyre szabott terápiás megközelítést tesz lehetővé. Ilyen esetben is a klinikus egyedileg tud dönteni arról, hogy mikor választ egy toxinkimutatást ELISA módszerrel, a székletből konvencionális tenyésztést vagy szofisztikáltabb genetikai markereket (12).
Az új generációs szekvenálási eljárások megválasztásánál is ezt kell figyelembe venni. Az S16 rRNS gén amplikonalapú szekvenálása során a kódoló génszakasz egy vagy néhány részletét vizsgálja. A teljes genom ún. shotgun- szekvenálása során pedig a teljes baktériumgenomot tárják fel. Utóbbi a virulencia-, toxin- stb. gének meghatározását is lehetővé teszi. A kérdés csak az, hogy a klinikai kérdésfeltevés és terápiás konzekvencia vonatkozásában ez többet nyújt-e. Ismertek-e egyáltalán az adott patogén dysbiosis hátterében azonosítható virulenciagének? Az adott labor ezeket képes-e detektálni, illetve az interpretációs csapat rendelkezik-e ezzel kapcsolatos klinikai tapasztalattal? Az amplikonszekvenálás magának a potenciális patogén ágens kimutatásában robusztusabb, és nagyobb publikált tapasztalat van vele. Megfontolandó, hogy – a Helicobacter-példánál maradva – ha a kórokozó azonosítása megtörtént, utána szükség szerint célzottan ugyanakkor egyedi géneket keressünk és vizsgáljunk az arra legérzékenyebb és legspecifikusabb módszerrel.
A mikrobiommal kapcsolatos (klinikai) közleményekben kevés szó esik a mintavételről. Mint minden laborvizsgálatnál, itt is döntő szerepe van annak, hogy a mintavétel antibiotikus kezelés vagy probiotikum után, egy új gyógyszer bevezetése előtt vagy után, esetleg specifikus diéta alatt történt-e, és megfelelő technikával, mintavevő kittel, megfelelő mennyiségben. A Helicobacter-példánál maradva a klinikum szempontjából fontosabb lehet, hogy honnan és hány mintát vesz az endoszkópos a szövettanra, mint hogy hematoxilin-eozin-, immun- vagy FISH-vizsgálat készül.
Egy másik fontos dilemma a betegségek mikrobiomasszociációjával kapcsolatban, hogy a patogén dysbiosisok nem csupán bakteriális, de gombás és virális eredetűek is lehetnek, amelyekről egyelőre nagyon keveset tudunk. A humán mikrobiom elkészült „atlaszának” ezek nem képezik részét. A humán szervezetben élő vírusok összessége, a „viriom” a bakteriális mikrobiomhoz hasonlóan rengeteg egyedi vírusból áll, amelyek diverzitása ezres nagyságrendű minden emberben, és szintén nagyfokú egyénre jellemző mintázatot ad. Folyamatban van a mikrobiomatlaszhoz hasonló feltérképezése a viriomnak is több anatómiai lokalizációban, de az eddig azonosított vírusok többségéről nagyon kevés biológiai információ áll rendelkezésre. Az NCBI adatbázis több mint tízezer vírus-
szekvencia adatait tartalmazza, amely a teljes biodiverzitás töredéke a mai tudásunk szerint (13). A humán viriom többsége a becslés szerint nem patogén jelentőségű, hanem mind a prokarióta sejteket fertőző (szabályozó) bakteriofág, illetve humán sejteket fertőzni képes eukarióta vírus. Ennek óriási a jelentősége, hiszen a bakteriális mikrobiom összetételének szabályozását, illetve a patogén baktériumok elszaporodását, magának a patogén dysbiosis kialakulását gátolhatják. Másrészt a vírusellenes kezelések fűnyíróelv szerint ezeket is károsítani tudják, ami épp az ökoszisztéma szabályozott egyensúlyának felborulását eredményezheti (14).
Az amplikon-, illetve a shotgunszekvenálás
Az új generációs szekvenálási módszerek használata lehetővé teszi egyetlen futtatás során a mikrobiális közösség teljes összetételének egyidejű vizsgálatát, ide értve az ún. nehezen tenyészthető mikroorganizmusok kimutatását és azonosítását. Az NGS jelentősége, hogy olyan esetekben is lehetővé teszi a mikroorganizmusok kimutatását, amelyeknek egyébként speciális növekedési feltételekre van szükségük, amelyek nem, vagy csak nagyon nehezen biztosíthatók laboratóriumi környezetben. Az NGS térhódítása előtt használt ún. PCR módszerhez előre kellett definiálni a vizsgálni kívánt taxonokat, annak érdekében, hogy kimutathatóak legyenek, így a mintában váratlanul előforduló baktériumokat nem detektálták.
Az amplikonszekvenálás és a shotgunszekvenálás egyaránt a nukleinsavak mintából történő extrakciójával kezdődik. A kivont DNS-ből az amplikonszekvenálás esetében a 16S rRNS gén célzott szakaszait specifikus, ún. primer párokkal kijelölve sokszorosítjuk. A fajazonosítás lényege, hogy a 16S (riboszóma) RNS-t kódoló gén annyira konzervált evolúciós értelemben, hogy variabilitása valós evolúciós távolságot jelez, amely a filogenetikai rokonság közelségével-távolságával korrelál (15).
1. táblázat: Az amplikon- és shotgunszekvenálás összehasonlítása
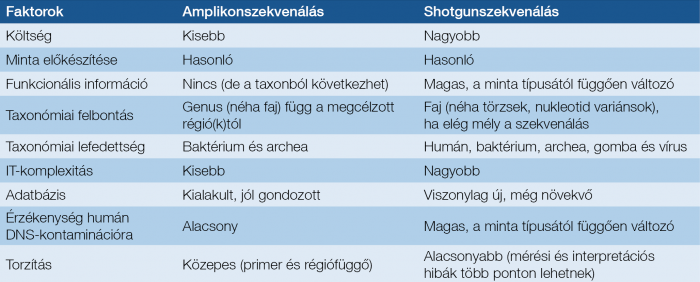
Fontos limitációja a módszernek, hogy az azonosítást szolgáló hipervariábilis szakaszokat az elérhető kitek nem fedik le teljesen, ezért eltérő taxonómiai eredményt adhatnak (16). Ezért a különböző laboratóriumok más és más metodikákkal készült mikrobiomleletei egymással nem, vagy csak korlátozottan összevethetők.
Az amplikonszekvenálás esetében baktériumfajok azonosítására ugyanaz a 16S rRNS génszakasz szolgál. Genetikai szerkezete 9 erősen konzervált régióból áll, amelyek univerzális primer kötőhelyként szolgálnak a PCR-amplifikációhoz, és 9 hipervariábilis régióból, amelyek az azonosításhoz használt jelentős szekvenciadiverzitást tartalmazzák. Az azonosítás során a kapott hipervariábilis régió összehasonlításra kerül olyan nagy, nyilvános adatbázisok 16S rRNS szekvenciáival, mint a SILVA, az NCBI, a Greengenes stb. Fontos azonban, hogy a taxonómiai azonosítás pontossága nagyban függ a használt adatbázis minőségétől és teljességétől. Továbbá a jelenleg leggyakrabban használt profilalkotási stratégia, a részleges 16S rRNS gének szekvenálása gyakran nem képes a baktériumok fajszintű taxonómiai megkülönböztetésére, így ez a módszer általában a nemzetségszintű osztályozásra korlátozódik.
Az ún. shotgun módszer esetében kis DNS-fragmenseket hoznak létre a kivont DNS-ből, és ezt közvetlenül szekvenálják válogatás nélkül, majd bioinformatikai algoritmusok segítségével megfeleltetik egy nagy genetikai adatbank adataival, és ez lehetővé teszi nemcsak az S16 szakasz azonosítását, de bármely tetszőleges funkcióval bíró gén azonosítását is.
Utóbbi a génszekvenciák funkcionális annotációja, amely két lépésből áll: az első lépés a génpredikció, amely során meghatározzák, mely szekvencia kódolhat (részben) fehérjéket; a második lépés a génannotáció, tehát a génpredikció során azonosított szekvenciákat összehasonlítják a fehérjecsaládok adatbázisával és annotálják a megfelelő család funkciójával. Azonban a génszekvenciák funkcionális annotációja arról nem nyújt információt, hogy a prediktált gének valóban kifejeződnek-e a vizsgált mintában. Mivel a shotgun-metagenomika PCR-független, detektálhat akár olyan ritka mikroorganizmusokat, amelyek elkerülhetik a célzott amplikonalapú NGS-módszerekkel való kimutatást az rRNS génen belüli self-splicingnek köszönhetően (17).
A shotgunszekvenálási módszer képes a mikroorganizmusok alacsonyabb taxonómiai szinteken való, faj- vagy akár törzsszintű megkülönböztetésére is. Fontos megjegyezni, hogy a mikroorganizmusok genomjának rekonstruálása a kis DNS-fragmensek keverékéből nagyon komplex, a szekvenálás felbontásnak optimalizálásához további bioinformatikai fejlesztést igénylő folyamat. Különösen a törzsszintű meghatározás folyamán az összeállítási algoritmusok számára kihívást jelenthetnek az ismétlődő elemek megfelelő beépítése, illetve az olyan kis genetikai különbségek, amelyeket nehéz megkülönböztetni a tényleges szekvenálási hibáktól (18).
Mind az amplikonszekvenálási, mind a shotgunszekvenálási módszernek vannak előnyei és hátrányai, a módszert érdemes annak függvényében kiválasztanunk, hogy mit várunk a vizsgálattól. Az amplikonszekvenálás gyorsabb, kevésbé komplikált és költséghatékonyabb, ezáltal könnyebben beilleszthető a klinikai rutinba, illetve míg a shotgunszekvenáláshoz használt adatbázisok még relatíve újak, az amplikonszekvenáláshoz használtak már kidolgozottak. Ugyanakkor az amplikonszekvenálás során – a shotgunmódszerrel ellentétben – esély van az amplifikációból származó adattorzulásra, illetve nem alkalmas a megcélzott géneken kívül más gének, illetve gombák, vírusok kimutatására.
A shotgunszekvenálási módszer tehát alkalmas mélyebb, akár törzsszintű taxonómiai felbontásra, révén elérhető a mintában található génekre vonatkozó információ, gombák és vírusok is kimutathatóak, lehetővé teszi a genomok de novo összeállítását, ugyanakkor hátránya, hogy magas readszámot igényel, a klasszifikációhoz referenciagenom szükséges, és érzékenyebb a host DNS kontaminációra (utóbbi a szekvenálási mélység kalibrálásával mérsékelhető) (19, 20).
A mikrobiomlelet értelmezése: a diverzitás és a dysbiosis
Egy komplex ökoszisztéma leírása statisztikai módszerekkel lehetséges, ami az orvosi rutingyakorlatban nem szokványos fogalmak bevezetését teszi szükségessé. Ezeket részleteiben kidolgozták a XX. században az ökológiai és evolúcióbiológiai kutatások során, ezért a módszertana és a matematikai háttere, valamint alkalmazása és limitációi jól ismertek. A gyakorló orvos számára mindazonáltal teljesen ismeretlen területet jelentenek, és mint minden statisztikai módszer, az alkalmazása és annak relevanciája függ a kérdésfeltevéstől, amire választ várunk. A teljesség igénye nélkül itt az alapfogalmak tisztázására vállalkozunk.
Egy ökoszisztéma jellemzésének alapja annak fajösszetétele, más néven fajgazdagsága vagy diverzitása. Egy adott populáció diverzitásának matematikai leírása az alfa-diverzitás. Ez a fajok számának megadása mellett lehetővé teszi a fentiek szerinti bonyolultabb statisztikai elemzéseket, amelyek előnyeit és limitációját az adott módszertan határozza meg. Alkalmas lehet a populáció „egészségének”, illetve potenciális sérülésének leírására is. Az esőerdők pusztulásának egyik fontos mérőszáma az ott élő élővilág fajgazdagságának csökkenése. Sokkoló adat a föld biodiverzitásának csökkenésére, hogy az emlősöknek csupán 4%-át adják a vadon élő állatok, 60%-át a tenyészállatok, 36%-át pedig az emberi faj. Ehhez hasonlóan a madarak 70%-a haszonállatként nevelt szárnyas (21). A fajok eltűnése, kihalása mellett tehát nagy jelentősége van a megoszlásnak is – nevezetesen hogy egy-egy faj milyen mértékben reprezentált.
A mindennapi gyakorlatban legtöbbet megjelenő index a Simpson-score. Ez annak a valószínűségét jelöli, hogy egy véges számú egyedből álló populációból véletlenszerűen kiemelt 2 egyed azonos taxonhoz tartozó lesz-e. Ez a taxonok relatív reprezentációjának (abundancia) súlyozott átlagát jelöli tulajdonképpen. Értéke 0 (tökéletesen diverz) és 1 (teljesen egynemű) között változhat, és egy valószínűségi értéket reprezentál. Miután a gyakorlatban a magasabb számhoz nagyobb diverzitás könnyebben érthető, ennek inverz értékét szokták megadni, ahol az 1-hez közelítve kapjuk a legdiverzebb, a nullához pedig a legkevésbé diverz populációt (22).
A Shannon-index ugyanakkor a megoszlás egyenletességét jelöli (evenness). Fontos limitációját jelenti, hogy feltételezi, hogy a mintában az összes taxon reprezentált (a minta elemszámát nem veszi figyelembe). Ha ez nem teljesül, a prediktív értéke jelentősen csökken, a vizsgált minta elemszámának csökkenésével (23).
1. ábra: A diverzitásszámítási paraméterek illusztrálása

Az ún. béta-diverzitás különböző populációk összehasonlításán alapul (pl. a budai és a pilisi természetvédelmi területen két hasonló fekvésű és méretű rét biodiverzitásának összehasonlítása, vagy egypetéjű, de más közegben élő ikerpár vastagbélflórájának összehasonlítása). Limitációját elsősorban a vizsgált populációk kiválasztása és a kérdés szempontjából releváns „összevethetősége” adja. A különbözőség ugyanis lehet annyira multifaktoriális, hogy a vizsgált tényező hatása ebből a szempontból elenyésző lehet. A mikrobiomdiagnosztikában ez egy sarkalatos tényező, ugyanis más etnikai hátterű, illetve más fizikai és szociális közegben élő emberek összehasonlítása nem feltétlenül ad választ egy külső behatás, pl. antibiotikus kezelés hatásának vizsgálata során. A gamma-diverzitás ugyanakkor a különböző ökoszisztémák összességének leírása, pl. a humán mikrobiomatlasz.
A gyakorlati alkalmazást segíti a diverzitásmutató percentilis skálán történő megjelenítése, ami, megint, kellően reprezentatív referencia-adatbázissal a birtokunkban az egyedre jellemző értékek összevetését segíti (24).
A funkcionális taxonómia
A fajok filogenetikai osztályozásán túlmutat a funkció szerinti csoportosítás. Ennek lehetőségei szinte beláthatatlanok, de a jelenségek komplexitása a gyakorlati alkalmazását erősen limitálja. A mikrobiomleleteken megjelenő F/B arány egy ilyen mutató, amely a propionát- és butiráttermelő taxonok egymáshoz viszonyított aránya révén bír (újabban már megkérdőjelezett [25]) prediktív értékkel az anyagcsere-betegségek, valamint a gyulladásos folyamatok kialakulása tekintetében. A mikrobiomdiagnosztika hőskorában definiált érték lényege, hogy a Firmicutes és Bacteroides törzsek egymáshoz viszonyított aránya korrelációt mutatott az elhízás, a diabétesz és a kardiovaszkuláris kockázat alakulásával (26).
Limitációját ugyanakkor egyrészt az adja, hogy számos olyan taxon torzítja a képlet számértékét, amely ugyan független kockázati tényezőként megjelenhet ezekben az anyagcsere-rendellenességekben (Prevotella család), ugyanakkor taxonómiailag a Bacteroides törzsbe tartoznak, tehát matematikailag az arányuk növekedése csökkenti az arányszám értékét. Miután szerepük nem obligát patogén, tehát intakt barrier mellett gyulladáskeltő hatásuk limitált, mégis a kellően nagy elemszámú klinikai tanulmányok statisztikailag szignifikánsnak mutatták a vizsgált populációk vonatkozásában a mérőszámot. Az individuális kockázatelemzés során azonban ezt nagyon is figyelembe kell venni, mert gyakran ad fals negatív értéket.
A limitáció másik tényezője magának a jelenségnek, a propionáttúlsúlynak a kóroki szerepe. A mikrobiommal foglalkozó tudományos ismeretterjesztő leírások visszatérő fordulata a „jó” és a „rossz” baktériumok aránya. Ez is az F/B arányhoz vezethető vissza, ami az első származtatott „mikrobiombiomarkerek” egyike volt, még jóval az új generációs szekvenálási eljárások elterjedése és a humánmikrobiom-projekt publikálása előtt. Ez az egyszerűsítés egy magas kockázatú betegpopuláció vizsgálata során prediktív értéket mutatott, azonban tudni kell, hogy a magas propionáttermelés és magának az ebből következő „inzulinrezisztenciának” (27) fontos evolúciós jelentősége volt, és önmagában egyáltalán nem kóros jelenség. Az emberi faj a mai tudásunk szerint konkrétan ezért élte túl az utolsó jégkorszakot. A medvék életciklusában ennek a jelentősége jól megérthető. Nyáron, amikor táplálék bőven áll rendelkezésre, a magas propionáttúlsúly gátolja, hogy a keringő glükóz az izomszövetben hasznosuljon. Az inzulinreceptor mTORc1 mediált blokkolásának evolúciós szerepe van, hogy ez a zsírszövetbe tudjon kerülni, és tárolódjon télire, amikor nem áll rendelkezésre a megfelelő mennyiségű élelem. Télen ugyanakkor, amikor a szervezet át tud állni a lelassult anyagcserére, csökkent agyi funkciók (téli álom) mellett a ketogén anyagcsere hetekig, hónapokig elegendő táplálékot biztosít a koplalás ideje alatt. Ezek a baktériumok és maga a propionáttermelés csak attól válik kórossá, hogy sem a zsírszövet hőtermelése, sem a télen-nyáron egyformán elérhető folyamatos, bőséges táplálékellátás nem teszi lehetővé ennek a tárolt zsírnak a felhasználását. Ez utóbbi valójában a kóros jelenség, és ennek oka nem a propionáttermelés, hanem a fiziológiás hasznosítás hiánya. Evolúciós szempontból nézve tehát, vagy állandóan magasabb butiráttermelést kell indukálnunk, vagy a propionáttúlsúly esetén ügyelni kell a hőleadásra és az időszakos koplalás beiktatására. Nem meglepő módon mindkét intervenció hatása klinikai vizsgálatokban is az inzulinrezisztencia reverzióját és fogyást, illetve az alacsonyabb inzulinszint mellett a diabétesz/elhízás kockázatcsökkenését eredményezte (28).
A szekvenciaadatok értékelése
Mivel a mikrobiomvizsgálatok hatalmas mennyiségű adatot termelnek, ezek elemzése informatikai eszközökkel lehetséges. A jelenlegi amplikonszekvenálás leggyakrabban régióspecifikus (V1-V3 vagy V4) ún. „forward” és „reverse” primereket használ, és az Illumina MiSeq technológia által támogatott nukleinsavhosszra támaszkodik (2×300 bp), hogy lefedhesse a lehető legtöbb variábilis régiót, illetve a lehető legpontosabb legyen.
Az amplikonszekvenálásból nyert adatok taxonómiai adatbázisok segítségével elemezhetőek. A taxonómia hozzárendelhető a szekvenciaadatokhoz olyan machine learning módszerekkel, mint a Ribosomal Database Project (RDP) Classifier, amely gyorsan és pontosan besorolja a bakteriális 16S rRNS szekvenciákat a Bergey-féle bakteriális rendszertan szerint, alkalmas egyetlen rRNS-szekvencia, vagy akár több ezer szekvenciát tartalmazó könyvtár elemzésére is (29). A taxonómiai hozzárendelés másik módja a népszerű referencia-adatbázisok használata, ilyen a Greengenes és a SILVA, amelyek a hozzárendeléshez szükséges felületet különböző mikrobiomanalízis-csomagok révén biztosítják. Ilyen például a mothur, amely egy számos korábbi eszközön alapuló, átfogó szoftvercsomag (30), a PyCogent toolkit felhasználásával készült „quantitative insights into microbial ecology” (QIIME) (31), illetve a DADA kiterjesztett és javított algoritmusú változata, a DADA2, amely modellezi és javítja az Illumina-szekvencia amplikonhibáit (32).
A shotgunszekvenálási adatok legjobb újjáépítési módszerével kapcsolatban még nincs konszenzus. Ez történhet de novo, referenciagenomok alapján, amely során gyakran a de Bruijn-gráf megközelítést használják, amely előnye a hagyományos OLC- (overlap layout consensus) gráfokkal szemben az NGS-adatok redundanciájának kezelésében és kihasználásában rejlik (33). De novo újjáépítéshez gyakran használják a MetaVelvet, az IDBAUD, a metaSPAdes és a MEGAHIT szoftvereket.
A referenciavezérelt metagenomikus újjáépítésben (pl. MetaCompass) a kontigok rekonstruálásához a szekvenálási adatokat referencia-adatbázisokhoz rendelik, ennek a módszernek a kritikus pontja a referenciagenomok elérhetősége és az adatbázisok minősége. Gyakran a taxonómiai hozzárendeléshez vagy génannotációhoz az újjá nem épített metagenomikai adatokból reprezentatív vagy megkülönböztető gének readalapú profilozását végzik a referencia-adatbázissal összevetve azokat. A taxonómiai tipizálás során hasznosak lehetnek a hasonló DNS-összetételek vagy nukleotidmintázatok (például egyedi k-mer-eloszlások), GC-tartalom vagy génhomológia. Ezzel szemben a MetaPhlAn2 például egyedi, kládspecifikus markergéneket használ a mikrobiális taxonok megkülönböztetéséhez (34).
A mikrobiom taxonómiai összetétele mellett azonosítani lehet a különböző mikrobiális populációk közötti funkcióbeli különbségeket. A 16S-szekvenálási adatokból, a közösségen belüli taxonok relatív eloszlását felhasználva megjósolható egy funkcionális profil olyan programokkal, mint a PICRUSt vagy a Tax4Fun. Ezek a programok megjósolhatják a géntartalom potenciális funkcionalitását az egyes jelen lévő taxonok referenciagenomja alapján. Az így nyert adatok megközelítések, hiszen nem veszik figyelembe a tényleges fehérjeexpressziót, és nagyban függnek a referenciagenomoktól és annotációiktól.
A shotgunszekvenálás eredményei is lehetővé teszik a funkcionális analízist: a metagenom újjáépítése után olyan eszközök végeznek génpredikciókat, mint például a MetaGeneMark és a Glimmer-MG. A kódoló gén azonosítása után funkcionális annotációt végeznek fehérjeszekvenia-homológián alapuló keresésekkel (UBLAST- és USEARCH-alapúak) ortológ (pl. EggNOG), enzim- (KEGG) vagy fehérjedomén-adatbázisokkal (Pfam, TIGRFAMs, InterPro) szemben.
A legnagyobb kihívást a leletek értékelésében ez a metodikai heterogenitás és az abból fakadó interpretációs különbségek jelentik. Maguk a szekvenciamérések génekkel történő beazonosítását végző szoftverek, illetve a mérés módszertanának különbözősége kódolja az eltérő értelmezést, akárcsak egy Celsius- vagy Fahrenheit-hőmérő esetén, csak több száz változó paraméter és származtatott statisztikai mérőszám révén. Ez az oka annak, hogy az egyes laborok adatai nem összevethetők a reportok alapján, csak az eredeti szekvenciaadatok újraértékelése révén. Ennek megfelelően a mérést végző labor módszertanával az interpretációt végző szakembernek vagy teamnek tisztában kell lennie, illetve gyakorlatot kell szereznie azok differenciáldiagnosztikai pontosságában, limitációiban egyaránt. A származtatott statisztikai módszerek összehasonlítása is problémás ennek megfelelően, hiszen a mikrobiomadatok potenciálisan több ezer taxonómiai egységet tartalmazhatnak, illetve különbségek vannak a szekvenálási mélységben is (35, 36).
A mikrobiomvizsgálat indikációja, a mikrobiomteamek és a betegtájékoztatás szerepe
A klinikai gyakorlatban a mikrobiomdiagnosztika első lépése a vizsgálat indikációjának meghatározása, amelyet (ma Magyarországon) leggyakrabban gasztroenterológus szakorvos kezdeményez. Nemzetközi szinten ugyanakkor már ma is sokkal szélesebb a paletta, és főként neurológiai, pszichiátriai, onkológiai és immunológiai területen kezd elterjedni a napi klinikai gyakorlatban (37).
A mikrobiomsérülés jellegéből fakadóan a kivizsgálás a patogén „góc” azonosítását célozza meg, illetve azokat a vizsgálatokat, amelyek a biológiai barrierek állapotára adnak információt. Ez utóbbiak definiálják a gazdaszervezet-mikrobiom interakció jellegét és intenzitását. Természetesen szükség van emellett azokra a specifikus labor-, képalkotó és egyéb rutinvizsgálatokra, amelyek a beteg klinikuma által kijelölt terápiás döntéshozatalt segítik. A kiindulópont azonban minden esetben a konkrét klinikai probléma és annak kóroktana, valamint terápiás opciói. Minden, a betegen végzett diagnosztika célja a terápiás opciók közötti választás, ezért a differenciáldiagnosztikai kérdés feltevését is ezek a terápiás opciók kell hogy vezessék.
A megfelelően és a megfelelő időben vett minta elemzése után az interpretáció már interdiszciplináris munka, amelyben a mikrobiomtesztek értékelésében járatos gasztroenterológus, molekuláris biológus, mikrobiológus, dietetikus, illetve egyéb klinikai szakorvos is (pl. neurológus, onkológus, immunológus stb.) részt vesz. A team feladata a terápiás terv összeállítása – az esetleges antibiotikus kezelés, a táplálásterápia tervezése vagy (ritka esetben) széklet-transzplantáció engedélyeztetése. A megvalósításban kiemelt szerepe van a regenerációs mikrobiomdiétában jártas dietetikusnak.
A mikrobiomkutatás óriási információs robbanást hozott, és hoz ma is naponta a medicinában, amit gyakorló orvosként szinte lehetetlen naprakészen tartani. A beteg gyógyításáért felelős szakorvosként azonban mód van a ma már hazánkban is elindult diagnosztikai laborokkal partnerségben ezeket az interdiszciplináris teameket kialakítani, ahol a szükséges emberi és technológiai kapacitások elérhetők.
Fontos ugyanakkor tájékoztatunk a betegeket, hogy a saját olvasmányélményei alapján az interneten „tesztek formájában vásárolt remény” kis eséllyel hozza meg a várt eredményt. Mind a tesztek indikációját, mind a kiértékelést az adott betegség kezelésében jártas szakorvosnak kell összefognia, és ennek kiindulópontja a beteg klinikai problémája kell hogy legyen. Ökölszabályként elmondható, hogy a mikrobiomteamben a beteg kezeléséért az a klinikai diszciplína felel elsődlegesen, amelyik a kezelési rutinprotokollok keretében az ellátást végezné. Az új kóroktani és patofiziológiai szemlélet, molekuláris biológiai tudás ezt az evidenciákon alapuló tapasztalatot egészíti ki.
A mikrobiominterpretációt ennek ismeretében és a kezelőorvossal teammunka keretében kell elvégezni, és a szolgáltatások árazásában az onkológiai (nemzetközi) gyakorlatnak megfelelően megjeleníteni (38). Ahogy a daganatos megbetegedések esetében a diagnosztika és a kezelés egyre komplexebb feladattá vált, interdiszciplináris teamek vették át a diagnosztika és a terápiatervezés komplex feladatát. A megoldás az onkoteam létrehozása volt, ami képessé tette a beteg ellátásában szerepet játszó orvoscsoportot a feladat leghatékonyabb végrehajtására.
Minden komplexitás és szervezési feladat ellenére a molekuláris medicina valós forradalmat hozott az ezredfordulót követő évtizedekben, és ez a folyamat nem lassulni, hanem tovább gyorsulni látszik. Az internet által biztosított konstans nyilvánosság megváltoztatja a klasszikus orvos-beteg viszonyt, ha tetszik, ha nem. A betegek információéhségét ki kell szolgálnunk, nem megtiltani, hogy tájékozódjanak az interneten. A tájékoztatás fárasztó, idő- és költségigényes feladat, de csak ez biztosíthatja a páciensek együttműködését a felkészült szakmai teamekkel, ami végső soron mindannyiunk érdeke.
Irodalom
2. Bland EF. Rheumatic fever: the way it was. Circulation 1987 Dec; 76(6): 1190–5.
https://doi.org/10.1161/01.cir.76.6.1190. PMID: 3315293.
3. Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. (Letter). Lancet 1983; 1: 1273–5.
4. Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter 2014 Sep; 19(Suppl 1): 1–5.
https://doi.org/10.1111/hel.12165. PMID: 25167938.
5. Roberts PJ. Human genome project. Annales chirurgiae et gynaecologiae 2001; 90(1): 3.
6. Clarridge JE 3rd. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev 2004 Oct; 17(4): 840–62. table of contents. https://doi.org/10.1128/CMR.17.4.840-862.2004.
PMID: 15489351; PMCID: PMC523561.
7. Hugenholtz P, Chuvochina M, Oren A, Parks DH, Soo RM. Prokaryotic taxonomy and nomenclature in the age of big sequence data. ISME J 2021 Jul; 15(7): 1879–1892. https://doi.org/10.1038/s41396-021-00941-x. Epub 2021 Apr 6. PMID: 33824426; PMCID: PMC8245423.
8. Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E, Weinstock GM. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun 2019 Nov 6; 10(1): 5029. https://doi.org/10.1038/s41467-019-13036-1. PMID: 31695033; PMCID: PMC6834636.
9. Varkondi E, Pinter F, Robert K, Schwab R, Breza N, Orfi L, Keri G, Petak I. Biochemical assay-based selectivity profiling of clinically relevant kinase inhibitors on mutant forms of EGF receptor. J Recept Signal Transduct Res 2008; 28(3): 295–306. https://doi.org/10.1080/10799890802084671. PMID: 18569529.
10. Akeel M, Elhafey A, Shehata A, Elmakki E, Aboshouk T, Ageely H, Mahfouz MS. Efficacy of immunohistochemical staining in detecting Helicobacter pylori in Saudi patients with minimal and atypical infection. Eur J Histochem 2021 Jul 20; 65(3): 3222. https://doi.org/10.4081/ejh.2021.3222. PMID: 34284564; PMCID: PMC8314390.
11. Mégraud F, Lehours P. Helicobacter pylori detection and antimicrobial susceptibility testing. Clin Microbiol Rev 2007 Apr; 20(2): 280–322. https://doi.org/10.1128/CMR.00033-06. PMID: 17428887; PMCID: PMC1865594.
12. Burnham CA, Carroll KC. Diagnosis of Clostridium difficile infection: an ongoing conundrum for clinicians and for clinical laboratories. Clin Microbiol Rev 2013 Jul; 26(3): 604–30. https://doi.org/10.1128/CMR.00016-13. PMID: 23824374; PMCID: PMC3719497.
13. Liang G, Bushman FD. The human virome: assembly, composition and host interactions. Nat Rev Microbiol 2021 Aug; 19(8): 514–527. https://doi.org/10.1038/s41579-021-00536-5. Epub 2021 Mar 30. PMID: 33785903; PMCID: PMC8008777.
14. Bai GH, Lin SC, Hsu YH, Chen SY. The Human Virome: Viral Metagenomics, Relations with Human Diseases, and Therapeutic Applications. Viruses 2022 Jan 28; 14(2): 278. https://doi.org/10.3390/v14020278. PMID: 35215871; PMCID: PMC8876576.
15. Rausch P, Rühlemann M, Hermes BM, Doms S, Dagan T, Dierking K, Domin H, Fraune S, von Frieling J, Hentschel U, Heinsen FA, Höppner M, Jahn MT, Jaspers C, Kissoyan KAB, Langfeldt D, Rehman A, Reusch TBH, Roeder T, Schmitz RA, Schulenburg H, Soluch R, Sommer F, Stukenbrock E, Weiland-Bräuer N, Rosenstiel P, Franke A, Bosch T, Baines JF. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 2019 Sep 14; 7(1): 133. https://doi.org/10.1186/s40168-019-0743-1. PMID: 31521200; PMCID: PMC6744666.
16. Tremblay J, Singh K, Fern A, Kirton ES, He S, Woyke T, Lee J, Chen F, Dangl JL, Tringe SG. Primer and platform effects on 16S rRNA tag sequencing. Front Microbiol 2015; 6: 771.
17. Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, Wilkins MJ, Wrighton KC, Williams KH, Banfield JF. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 2015 Jul 9; 523(7559): 208–11. https://doi.org/10.1038/nature14486. Epub 2015 Jun 15.
18. Boers SA, Jansen R, Hays JP. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur J Clin Microbiol Infect Dis 2019 Jun; 38(6): 1059–1070. https://doi.org/10.1007/s10096-019-03520-3. Epub 2019 Mar 5. PMID: 30834996; PMCID: PMC6520317.
19. Di Segni A, Braun T, BenShoshan M, Farage Barhom S, Glick Saar E, Cesarkas K, Squires JE, Keller N, Haberman Y. Guided Protocol for Fecal Microbial Characterization by 16S rRNA-Amplicon Sequencing. J Vis Exp 2018 Mar 19; 133: 56845. https://doi.org/10.3791/56845. PMID: 29608151; PMCID: PMC5933208.
20. Breitwieser FP, Lu J, Salzberg SL. A review of methods and databases for metagenomic classification and assembly. Brief Bioinform 2019 Jul 19; 20(4): 1125–1136. https://doi.org/10.1093/bib/bbx120. PMID: 29028872; PMCID: PMC6781581.
21. https://www.theguardian.com/environment/2018/may/21/human-race-just-001-of-all-life-but-has-destroyed-over-80-of-wild-mammals-study
22. SIMPSON, E. Measurement of Diversity. Nature 1949; 163: 688. https://doi.org/10.1038/163688a0
23. https://cran.r-project.org/web/packages/tabula/vignettes/diversity.html
24. Gibbons SM, Duvallet C, Alm EJ. Correcting for batch effects in case-control microbiome studies. PLoS Comput Biol 2018 Apr 23; 14(4): e1006102. https://doi.org/10.1371/journal.pcbi.1006102. PMID: 29684016; PMCID: PMC5940237.
25. Magne F, Gotteland M, Gauthier L, Zazueta A, Pesoa S, Navarrete P, Balamurugan R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020 May 19; 12(5): 1474. https://doi.org/10.3390/nu12051474. PMID: 32438689; PMCID: PMC7285218.
26. Stojanov S, Berlec A, Štrukelj B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease. Microorganisms 2020 Nov 1; 8(11): 1715. https://doi.org/10.3390/microorganisms8111715. PMID: 33139627; PMCID: PMC7692443.
27. Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, Wu H, Carreras A, Jeong H, Olofsson LE, Bergh PO, Gerdes V, Hartstra A, de Brauw M, Perkins R, Nieuwdorp M, Bergström G, Bäckhed F. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018 Nov 1; 175(4): 947–961.e17. https://doi.org/10.1016/j.cell.2018.09.055. Epub 2018 Oct 25. PMID: 30401435.
28. Lee JH, Park A, Oh KJ, Lee SC, Kim WK, Bae KH. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int J Mol Sci 2019 Oct 4; 20(19): 4924. https://doi.org/10.3390/ijms20194924. PMID: 31590292; PMCID: PMC6801758.
29. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007 Aug; 73(16): 5261–7. https://doi.org/ 10.1128/AEM.00062-07. Epub 2007 Jun 22. PMID: 17586664; PMCID: PMC1950982.
30. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009 Dec; 75(23): 7537–41. https://doi.org/10.1128/AEM.01541-09. Epub 2009 Oct 2. PMID: 19801464; PMCID: PMC2786419.
31. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010 May; 7(5): 335–6. https://doi.org/10.1038/nmeth.f.303. Epub 2010 Apr 11. PMID: 20383131; PMCID: PMC3156573.
32. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 2016 Jul; 13(7): 581–3. https://doi.org/10.1038/nmeth.3869. Epub 2016 May 23. PMID: 27214047; PMCID: PMC4927377.
33. Compeau PE, Pevzner PA, Tesler G. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol 2011 Nov 8; 29(11): 987–91. https://doi.org/10.1038/nbt.2023. PMID: 22068540; PMCID: PMC5531759.
34. Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 2015 Oct; 12(10): 902–3. https://doi.org/10.1038/nmeth.3589. Erratum in: Nat Methods 2016 Jan; 13(1): 101. PMID: 26418763.
35. Gao B, Chi L, Zhu Y, Shi X, Tu P, Li B, Yin J, Gao N, Shen W, Schnabl B. An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules 2021 Apr 2; 11(4): 530. https://doi.org/10.3390/biom11040530. PMID: 33918473; PMCID: PMC8066849.
36. Galloway-Peña J, Hanson B. Tools for Analysis of the Microbiome. Dig Dis Sci 2020 Mar; 65(3): 674–685. https://doi.org/10.1007/s10620-020-06091-y. PMID: 32002757; PMCID: PMC7598837.
37. Jarecsny T, Trencséni D, Peták I, Mechtler L, Frecska E, Schwab R. A mikrobiom-asszociált gyulladás és a migrén: a bél–agy tengely újabb állomása. CEUJGH 2022; 8(1): 17–22. https://doi.org/10.33570/CEUJGH.8.1.17
38. De Ieso P, Coward J, Letsa I, et al. A study of the decision outcomes and financial costs of multidisciplinary team meetings (MDMs) in oncology. Br J Cancer 2013; 109: 2295–2300. https://doi.org/10.1038/bjc.2013.586