
Reviews
Metabolic and storage disorders of the liver in childhood
Summary
The lysosome is an intracellular organelle that is a key component in cellular recycling and homeostasis. Lysosomal storage disorders (LSDs) are a large group comprising of over 50 disorders with an estimated combined prevalence of approximately 1 in 8000. The fundamental pathological mechanism of these disorders is of a single enzyme deficiency leading to defective degradation or transport of a complex molecules. The consequence is a build-up of ‘storage’ material that impacts on normal cellular function and homeostasis. These conditions are commonly progressive and multisystemic, effecting systems to a highly variable degree. Musculoskeletal involvement can include hypotonia, dysostosis multiplex, cardiomyopathy. Endothelial reticular involvement can include hepatosplenomegaly and bone marrow dysfunction. Neurological involvement is common and is the most devastating feature of LSDs. Hepatomegaly, usually associated with splenomegaly, is a common feature of LSDs. Of all the LSDs, Gaucher disease, Niemann–Pick A, B and C are most commonly associated with liver dysfunction, and are considered in more detail below.
Összefoglalás
A lizoszómák olyan intracelluláris sejtalkotók, amelyek kulcsszerepet töltenek be a sejtek homeosztázisában és újrahasznosítási folyamataiban. A lizoszomális tárolási betegségek (LSD) közé több mint 50 kórkép tartozik, ezek együttes, vélt prevalenciája 1:8000. A patomechanizmus alapja az egyes komplex molekulák (pl. szfingolipid, mukopoliszacharid) károsodott lebontása vagy szállítása egy adott enzimdefektus következtében. Ennek következtében az intermedier anyagcseretermékek felszaporodnak, és károsítják a normális sejtműködést. Ezek a folyamatok gyakran progresszívek és multiszisztémásak, azaz több szervet is érintenek, de az érintettség mértéke igencsak különböző lehet. A muszkuloszkeletális érintettség következtében izomhipotónia, csontrendszeri eltérések, cardiomyopathia alakulhat ki. A reticuloendothelialis rendszer érintettsége esetén hepatosplenomegalia és csontvelői működészavar jelentkezhet. A legsúlyosabb tüneteket az idegrendszer érintettsége esetén láthatjuk. A legtípusosabb tünet a hepatomegalia, amelyet gyakran splenomegalia is kísér. A leggyakoribb LSD, amely májműködési zavarral társul, a Gaucher-kór, a Niemann–Pick (NP) A, B és C típusa, a Pompe-kór, valamint a lizoszomális savas lipázhiány, amelyeket részletesen tárgyalunk. A tárolási betegségek közül az alfa-1 antitripszinhiányt és a Wilson-kórt beszéljük meg.
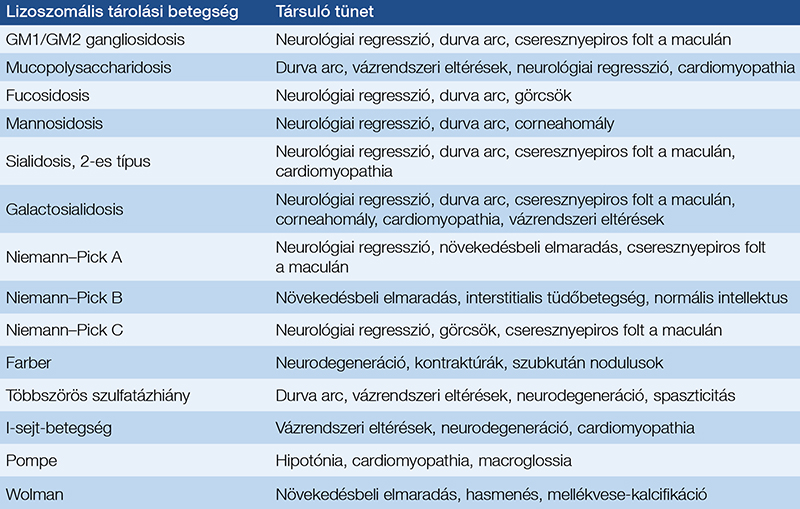
A hepatosplenomegaliával járó LSD-k listája az 1. táblázatban található.
Gaucher-kór
A Gaucher-kór (GD) a lizoszomális b-glükocerebrozidáz hiánya következtében kialakuló, autoszomális, recesszív módon öröklődő kórkép. Az enzimhiány következtében glükocerebrozid és egyéb glükolipidek halmozódnak fel az érintett szövetekben, így okozva szervkárosodást. Előfordulását a populációban 1:40 000-re teszik. A betegség klinikai megjelenése igen széles végletek közt mozog, kezdve a perinatális halállal járó súlyos formától az aszimptomatikus felnőttkori formáig. Klinikai osztályozás alapján 3 csoportot különítünk el. Az 1-es típus a krónikus, nem neuropátiás forma, a 2-es típus az akut neuropátiás forma és a 3-as a krónikus neuropátiás forma. A 2-es típus gyors progresszióval jár, és korai halálhoz vezet, míg a 3-as lassan progrediál, enyhébb tünetekkel kísérve. A betegek túlnyomó többsége az 1-es, nem neuropátiás formába tartozik, míg a 2-es típus kb. 1, a 3-as kb. 5%-ban fordul elő. A kórképet okozó GBA génnek több mint 300 mutációja ismert, de a betegek 50-60%-ában négy mutáció mutatható ki. A betegek többsége a fő prezentációs tünetek (splenomegalia, cytopenia) alapján általában hematológushoz kerül először. További figyelemfelkeltő tünet lehet a visszatérő csontfájdalom és a növekedésbeli elmaradás. A rekombináns enzimmel történő enzimpótló kezelés biztonságos és hatékony, ha a betegséget időben felismerik. Késői diagnózis esetén azonban egyes szövődmények (avaszkuláris csontnekrózis, máj-, lép- és csontvelőfibrózis) irreverzibilisek lehetnek, ezért a korai felismerés kulcsfontosságú (1).
Niemann–Pick A/B
A betegség lényege a szfingomielint bontó savanyú szfingomielináz (ASM) enzim hiánya. A hiányzó vagy csökkent formában működő ASM következtében szfingomielin szaporodik fel a szervezetben. A súlyosabb „A” típusban a szfingomielin a központi idegrendszerben halmozódik fel, így súlyos neurológiai tünetek fognak kialakulni, rossz életkilátással. A gyakoribb „B” típusban a szfingomielin a májban, lépben, tüdőben és a csontokban rakódik le. Ez is súlyos betegség lehet, de általában lassabb progresszióval jár. Az átmeneti formában, az „A/B” típusban kisfokú idegrendszeri érintettség mellett jelen vannak visceralis tünetek is.
Niemann–Pick C
A Niemann–Pick-betegség „C” típusa (NPC) a sejten belüli koleszterinszállítási zavar következtében alakul ki, és a nem észterifikált koleszterin lizoszómán belüli felhalmozódásával jár. A klasszikus megjelenési formája az újszülöttkori sárgaság és májműködési zavar, amelyek a későbbiekben oldódnak, de progresszív neurológiai károsodás alakul ki, általában 3-13 éves kor között. A diagnózis felállítása nehéz, mert az idegrendszeri tüneteket nem mindig előzi meg májérintettség, másrészről az újszülöttkori hepatikus tünetek jelentkezésekor még nem észlelhetőek neurológiai tünetek. A perzisztáló splenomegalia jellegzetes tünet, az idegrendszeri tünetek közül kiemelendő a vertikális tekintésbénulás és a gelasztikus cataplexia. A neurológiai tünetek általában progresszív lefolyást mutatnak, kezdve a hipotóniától a disztóniáig, dysphagiáig és pszichiátriai tünetekig. Ha a neurológiai tünetek korán jelentkeznek, az rossz prognózisra utal.
Újabban a szérum oxiszterolkoncentrációjának mérése segíti a diagnosztikát, emelkedett esetekben pedig a betegséget okozó NPC1 és NPC2 gének vizsgálata javasolt. A kezelés főleg tüneti a máj és idegrendszeri tünetek vonatkozásában, a szubsztrátredukciós készítmény célja az idegrendszeri tünetek progressziójának lassítása (1).
Pompe-kór
Az úgynevezett Pompe-betegség a ritka betegségek, pontosabban a genetikai hiba miatt öröklődő, sejten belüli anyagcserezavar okozta betegségek csoportjába tartozik. A savas alfa-glükozidáz teljes vagy részleges hiánya következtében a glikogén lebontása zavart szenved, és felhalmozódik a lizoszómákban minden szövetben. A kialakuló klinikai tünetekért az izomszövetben és a szívben felhalmozódó glikogén a felelős.
Lefolyás alapján három formát különböztetünk meg. Az első, az úgynevezett klasszikus, kora csecsemőkorban súlyos tünetekkel járó infantilis forma. Ezek a csecsemők az első hónapokban még normálisan fejlődnek, de 3-4 hónapos kor után azonban megjelennek az első tünetek: testszerte izomgyengeség tapasztalható, petyhüdtté válik az izomzat, nem tudják tartani magukat. Egyre gyengébbek lesznek, nehezen táplálhatók, súlygyarapodásuk lelassul, majd leáll, és végül fogyni kezdenek. Ez a folyamat kedvezőtlenül hat a légzésre és a szívműködésre is. Emiatt az amúgy is megnagyobbodott szív működése romlik, ez keringési zavarhoz, majd szívelégtelenséghez vezet. Eközben a csecsemő értelmi fejlődése mindvégig a kornak megfelelő. Sajnos az ebben a formában szenvedők többnyire nem érik meg a második életévüket, légzési és szívelégtelenségben veszítjük el őket.
Jóval enyhébb a kisgyermekkorban kezdődő, és a juvenilis-felnőttkori Pompe-betegség. A kisgyermekkori Pompe-betegség 15 éves kor előtt jelentkezik, többnyire fáradékonysággal, kimerültséggel. Jellemző tünete, hogy az érintett gyerekeket hamar kifárasztja a lépcsőn való járás, ügyetlenebbek, mint társaik, mozgásaik rendezetlenek, nem vesznek részt társaikkal a közös játékban, mert minden mozgás fárasztja őket. Náluk a szívizom megbetegedése csak késői tünet.
A juvenilis-felnőttkori forma esetében a betegeket lassan progrediáló izomsorvadás sújtja, járásuk egyre nehezebb, majd mozgásképtelenekké válnak. Légzési elégtelenség miatt kezdetben alkalmi, majd tartós légzéstámogatásra szorulnak. Hatékony terápia hiányában életkilátásaik igen szerények.
Szerencsére az utóbbi években, hosszas kutatások eredményeként lehetővé vált a Pompe-betegség kezelése intravénás, rekombináns humán alfa-glükozidáz enzim pótlásával (1).
Lizoszomális savanyúlipáz-deficiencia
A lizoszomális savanyúlipáz-deficiencia (LAL-D) egy ritka, autoszomális, recesszív módon öröklődő, lizoszomális tárolási betegség, amely jelentős koleszterin- és trigliceridfelhalmozódáshoz vezet a lizoszómákban. A lizoszomális savanyúlipáz-deficiencia azonos a gyermekekben régen leírt Wolman-betegséggel, illetve felnőttkori koleszterin-észter-tárolási betegséggel.
A LAL-D pontos előfordulása nem teljesen ismert, amelynek hátterében elsősorban a betegség aluldiagnosztizáltsága áll. A betegség prevalenciája 1:40 000 és 1:300 000 közé tehető.
A LAL-D-ben szenvedő betegek 86%-ában alakul ki májkárosodás. A hepatocytákban és a Kupffer-sejtekben felhalmozódó koleszterin és triglicerid microvesicularis steatosishoz, zsírmájhoz, hepatomegaliához, illetve emelkedett transzamináz- (elsősorban GPT-) értékekhez vezethet. A betegség előrehaladtával fibrózis, cirrózis és portális hipertónia alakul ki, amelyek végül májelégtelenséghez vezetnek. Kardiovaszkuláris manifesztáció a betegek 87%-át érinti. A szérumban az emelkedett LDL-koleszterin- és csökkent HDL-koleszterin-szintek, azaz a dyslipidaemia felgyorsult ateroszklerózist, koronáriabetegséget okoz, amely végül miokardiális infarktushoz és stroke-hoz vezethet.
A LAL-D splenomegaliához vagy egyéb, léppel kapcsolatos eltéréshez is vezethet a betegek 36%-ában. A lépben lerakódó zsírok és koleszterinek miatt a lép megnagyobbodik, ezért megnő a traumás lépruptúra veszélye, valamint splenectomiára is gyakrabban van szükség ezen betegek esetében. A hypersplenismus következtében a LAL-D-ben szenvedő betegek anémiásak, thrombocytopeniásak is lehetnek. Gasztrointesztinális manifesztáció ritkábban alakul ki, csupán a betegek 22%-ában figyelhető meg. A LAL-D gasztrointesztinális tünetei lehetnek a hasi, főként epigasztriális fájdalom, hányás, epehólyag-diszfunkció, diarrhoea, gasztrointesztinális vérzés vagy malabszorpció. Mindezek elmaradt növekedést, cachexiát, illetve alacsony növést eredményezhetnek.
A diagnózis felállítását elsősorban a szárított vércseppminta (dried blood spot, DBS) segítségével mérhető LAL-aktivitás adja meg. Ha enzimaktivitás nem detektálható vagy normálérték alatti, a LAL-D diagnózisa kimondható. A LAL-D egy életre szóló kezelést igénylő betegség, ezért a diagnózis megerősítése céljából a LIPA gén szekvenálása végezhető, amely a későbbiekben akár további prognosztikai információkkal is szolgálhat majd a rendelkezésre álló adatok növekedésével.
2015 óta elérhető a betegség enzimpótló kezelése, a sebelipáz-alfa, amely a LAL rekombináns formája (2).
A Wilson-kór gyermekkori vonatkozásai
A Wilson-kór (WD) egy autoszomális, recesszív módon öröklődő rézanyagcsere-zavar, amelynek hátterében egy ATP-áz (ATP7B) mutációja áll. Ennek következtében progresszív, toxikus rézfelhalmozódás alakul ki a szervezetben. Az ATP7B-nek több mint 500 mutációja ismert, közülük a leggyakoribb a H1069Q mutációja a kaukázusi populációban. A WD becsült incidenciája 1:30 000-re tehető. A kóros rézlerakódás már csecsemőkorban megkezdődik, kezdetben a májban, majd más szervekben is (idegrendszer, cornea, vese, szív). Ha a WD-t nem ismerik fel időben, végállapotú májelégtelenség, irreverzibilis idegrendszeri károsodás alakulhat ki.
A réz az epén keresztül ürül ki a szervezetből. A hepatocytába való felvételben, sejten belüli transzportban több molekula összehangolt működése szükséges (pl. a réztranszporter 1, metallothioneinek, metallochaperonok), amelyek lehetővé teszik az ATP7B fehérjével való kötődését. Az ATP7B protein fontos szerepet tölt be a réz apocöruloplazminnal való kötődésében, az effektív rézexkrécióban.
A rézakkumuláció az egyes szervekben különböző tüneteket okozhat. Gyermekkorban leggyakrabban a májérintettséggel találkozunk. A klinikai kép a tünetmentes transzamináz-emelkedéstől (>1 éves életkor) kezdve az akut hepatitisen, hepatomegalián, ultrahangon észlelt hiperechogén májszerkezeten, az akut májelégtelenségen át a végállapotú májcirrózisig terjedhet. Bár 5 éves életkor előtt ritkán okoz tüneteket, mégis 3 éves kor felett bármely életkorban megjelenhetnek a tünetek. A neurológiai/pszichiátriai tünetek jellemzően a második, harmadik évtizedben jelentkeznek, 10 éves kor előtt is csak extrém ritkán észlelhetünk neurológiai tüneteket. A gyermekkori, hepatikus kezdetű WD 4-6%-ában jelen van idegrendszeri érintettség is. Enyhe kognitív zavar (memóriazavar, nyelvi nehézség) azonban vélhetően ennél is gyakoribb. A corneában lerakódó réz okozta Kayser–Fleischer-gyűrű ritkán látható enyhe hepatikus tünetek mellett gyermekkorban, azonban gyermekkori neurológiai érintettség esetén szinte mindig jelen van. Akut hemolízis is lehet a WD prezentációs tünete, amely főleg fulmináns WD-hez társul, a prevalenciáját 6,9%-ra teszik.
A WD diagnózisának felállítása biokémiai, szövettani, genetikai teszteken, valamint az egyéb krónikus májbetegségek kizárásán alapul. WD-ben a szérumban többnyire <20 mg/dl alatti cöruloplazminszintet mérünk, amelynek hátterében a csökkent bioszintézis és a szabad apocöruloplazmin rövid féléletideje áll. Fontos azonban megjegyezni, hogy alacsonyabb szérum-cöruloplazminszintet észlelhetünk a heterozigóta hordozók akár 20%-ában, vagy egyéb eredetű májelégtelenségben, malabszorpcióban, glikozilációs zavarban, Menkes-betegségben, protein-energia malnutrícióban, nephrosis szindrómában, proteinvesztő enteropathiában, herediter acaeruloplasminaemiában.
Csecsemőkorban fiziológiásan is alacsonyabb szérum-cöruloplazminszintet detektálhatunk, majd a mennyisége növekszik egészen a pubertáskorban észlelt enyhe csökkenésig. Egyéves életkor alatt nem javasolt cöruloplazminszint-meghatározást végezni WD irányába.
WD-ben javasolt az elsőfokú rokonok szűrése is (szülők, testvérek, később leszármazottak) WD irányába, hiszen akár évtizedekig tünetmentes is lehet a rézanyagcsere zavara. A szűrés biokémiai mutatókon, valamint rézanyagcserét jellemző teszteken – lehetőség szerint célzott genetikai teszteken – alapul.
A WD kezelésének alapját a felhalmozódó réz eltávolítása adja. Ennek bázisát a kelátképzők vagy az intesztinális felszívódást gátló szerek jelentik. A táplálékkal bevitt réz megszorítása nem előzi meg a WD kialakulását, így ha tudomásunk van tünetmentes WD-s betegről (pl. családszűrésen igazolt testvér), nem elégséges rézszegény diéta tartása. Azonban az érintetteknek javasolt a rézszegény diéta, amíg az adekvát kezelés mellett a tünetek nem szűnnek, és a biokémiai eltérések nem normalizálódnak. A kezelést javasolt megkezdeni a diagnózis felállításakor tünetes betegeknél, míg tünetmentes, szűréssel diagnosztizált betegeknél 2-3 éves életkor felett. A jelenlegi ajánlás szerint tünetes betegek elsővonalbeli kezeléseként kelátképzők bevezetése javasolt (D-penicillamin, trientin). Arra vonatkozóan, hogy az effektív kelátképző kezelés után érdemes-e, mikor javasolt fenntartó kezelésre váltani, nincs egyértelmű ajánlás. Ennek megítélésében lehet segítségünkre a gondozás során a szérumréz és vizeletben a rézürítés követése. A tünetek, laboratóriumi eltérések (pl. emelkedett transzaminázok) normalizálódása hónapokat vehet igénybe, de előfordulhat irreverzibilis károsodás is (pl. neurológiai tünetek), amelyek csekély javulása várható (3).
Az alfa-1-antitripszin hiánya
Az α1-antitripszin hiánya (AATD) egy relatíve gyakori genetikai rendellenesség, gyakorisága 1:1600–1:2000 élveszülés. Az autoszomális kodomináns öröklődésmenetet mutató kórkép következtében az AAT szintje 85-90%-os csökkenést mutat a szérumban. A genetikai hiba következtében kialakuló mutáns AAT molekula abnormális foldingja alakul ki, amely miatt a fehérje „beragad” az endoplazmás reticulumba, és nem tud kiválasztódni a testnedvekbe és a vérbe. Az alfa-1-antitripszin-hiánybetegség változatos megjelenésének hátterében jelentős földrajzi különbségek, illetve a genetikai variációk állnak. Fiatalkorban jelentkező májbetegség és emphysema esetén gondolni kell erre a betegségre.
A főleg gyermekkorban jelentkező májbetegséget nem az alacsony szérumszint, hanem a hepatocytákban felhalmozódó AAT okozza, amely az endoplazmás reticulum stresszét, a májsejtek apoptosisát okozza (gain-of-function mechanizmus).
Ezzel ellentétben a fiatal felnőttkorban jelentkező tüdőbetegség kétségkívül az AAT alacsony szérumkoncentrációjával függ össze (loss-of-function mechanizmus).
Érdekes módon a PiZZ-mutációt hordozók csak 8-10%-ában alakul ki klinikailag súlyos májbetegség az élet első 20 évében, mégis ez a leggyakoribb genetikai májbetegség, amely miatt májátültetés történik gyermekkorban (4).
Csecsemőkorban leginkább elhúzódó sárgaság, neonatalis cholestasis hívhatja fel a figyelmet a kórképre. A laboratóriumi vizsgálatok mérsékelt transzamináz- (GOT-, GPT-, GGT-) emelkedést és direkt hyperbilirubinaemiát jelezhetnek. Ritkán gasztrointesztinális vagy köldökcsonkvérzés formájában is megnyilvánulhat a kórkép. Az irodalomban leírnak fulmináns májelégtelenség formájában induló formákat is (ascites, májszintetikus és alvadási zavar). Néhány esetben a betegeknél viszketés és hypercholesterinaemia is felléphet, amely miatt a biliaris atresiától való elkülönítés kiemelten fontos.
A csecsemőkoron túl a későbbiekben szinte bármikor okozhat hepatikus tüneteket az AATD. Elődomborodó has, hepatosplenomegalia, ascites vagy akár gasztrointesztinális vérzés is jelezheti a betegség előrehaladott voltát.
Felnőttkorban ismeretlen eredetű hepatopathia, krónikus hepatitis, portalis hypertensio, májcirrózis és hepatocelluláris karcinóma (HCC) hátterében keresni kell az AATD-t. Különösen nagy a jelentősége annak, hogy az AATD májcirrózis fennállása nélkül is fokozza a májrák kialakulásának kockázatát felnőttkorban.
Az AATD diagnózisa laboratóriumi módszerekkel felállítható. Mivel akutfázis-fehérje, gyulladás jelenléte esetén a szintje fals pozitívan lehet normális. Korábban a diagnózis „gold standarja” az izoelektromos fókuszálás volt. Manapság a genetikai vizsgálatok könnyebb elérhetőségével a SERPINA1 gén vizsgálatával is juthatunk diagnózishoz.
Májbiopszia elvégzése manapság már nem szükséges a diagnózis felállításához. Ha egyéb okok miatt történik, akkor a hepatocytákban felhalmozódó AAT molekulák jellegzetes festődési reakciót adnak PAS-festéssel, amelyek diasztáz emésztéssel rezisztensek.
Az AATD pulmonális szövődményeinek kezelésére rendelkezésre áll humán rekombináns AAT, míg a májtünetek kezelésére klinikai vizsgálatok zajlanak interferaló RNS-sel (fazirsiran) (5).
Irodalom
http://dx.doi.org/10.1017/CBO9781139012102
2. Vajda D, Szabó D, Dezsőfi A. Lizoszomális savanyú lipáz deficiencia. Gyermekgyógyászat 2019; 70(4): 225–229.
3. Socha P, Janczyk W, Dhawan A, et al. Wilson’s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for
Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2018 Feb; 66(2): 334–344.
https://doi.org/10.1097/MPG.0000000000001787 PMID: 29341979.
4. Sveger T. The natural history of liver disease in alpha-1-antitrypsin deficient children. Acta Paediatr Scand 1995; 77: 847–51
https://doi.org/10.1111/j.1651-2227.1988.tb10767.x
5. Strnad P, Mandorfer M, Choudhury G, et al. Fazirsiran for Liver Disease Associated with Alpha1-Antitrypsin Deficiency. N Engl J Med 2022 Aug 11; 387(6): 514–524. Epub 2022 Jun 25. PMID: 35748699.
https://doi.org/10.1056/NEJMoa2205416